
Introduction
The KlebNET-GSP Antimicrobial Resistance (AMR) Genotype-Phenotype Group collects matched genotype and phenotype data from the Klebsiella pneumoniae species complex isolated from diverse sources to test and improve tools for prediction of AMR from genomic data.
This initiative is led by Prof. Kathryn Holt and Dr. Kara Tsang and currently includes ~150 members across 44 research groups/projects, who have collectively shared >15,000 isolate data from ~35 countries, sampled from a variety of sources, including, humans, animals, and environments! The collective dataset also covers antibiogram data for 70 drugs (noting that not every isolate was tested against every antibiotic).
We plan to:
-
- Establish association of individual and combinatorial genotypes to AMR phenotypes in KpSC
- Implement findings in Kleborate for phenotype prediction that will be made available for use by other tools.
- Continuously assess the evidence and support for AMR genotype-phenotype relationships as additional data is accumulated.
- Study AMR prediction accuracy using (the updated) Kleborate and identify areas of remaining unexplained resistance.
- Apply genome wide association and machine learning methods to screen for potentially novel resistance determinants.
We have also joined forces with ESGEM-AMR Klebsiella pneumoniae Working Group due to our alignment in goals and expertise, with which we will curate/review AMRrules specific to KpSC.
First consortium outputs
Diversity, functional classification and genotyping of SHV β-lactamases in Klebsiella pneumoniae
In this first collaborative paper from the KlebNET AMR Genotype-Phenotype Group, matched genome and antimicrobial susceptibility data for 3,999 Klebsiella pneumoniae with only blaSHV beta-lactamases was analysed to assess genotype–phenotype relationships for blaSHV. SHV enzyme function associated with specific aa substitutions was explored and some SHV alleles were systematically assigned to functional classes based on the presence of these mutations. This resulted in 37 SHV alleles being re-classified compared with the current assignments in the NCBI’s Reference Gene Catalog and/or BLDB. Phylogenetic and comparative genomic analyses support that (i) SHV-1 (encoded by blaSHV-1) is the ancestral chromosomal variant, (ii) ESBL- and BLI-resistant variants have evolved multiple times through parallel substitution mutations, (iii) ESBL variants are mostly mobilized to plasmids and (iv) BLI-resistant variants mostly result from mutations in chromosomal blaSHV. The approach and results outlined here have been implemented in Kleborate v2.4.1, whereby known and novel blaSHV alleles are classified based on causative mutations. This study demonstrates the power of sharing AMR phenotypes alongside genome data to improve the understanding of resistance mechanisms.
Full paper and details here.

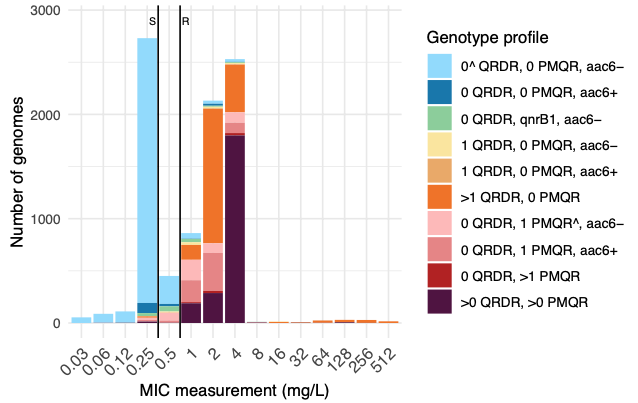
In this second collaborative paper from the group, we used 12,167 Klebsiella pneumoniae species complex (KpSC) genomes with matched ciprofloxacin susceptibility data, to explore the genetic basis for ciprofloxacin resistance. We developed a rules-based classifier to predict ciprofloxacin resistance by categorizing the number of quinolone resistance determining regions mutations in gyrA and parC, the number of plasmid-mediated quinolone resistance genes, and the presence/absence of aac(6′)-Ib-cr (which can acetylate ciprofloxacin). Predictive performance was assessed using the discovery dataset, for which we re-phenotyped discrepant isolates; and validated using an additional 7,0303 externally contributed isolates.
The rules-based classifier predicted R vs S/I with categorical agreement, sensitivity, and specificity >96%, and major/very major error rates <4%. Performance was similar across diverse KpSC sources (human, animal, other), species, and intra-species lineages. External validation of the classifier yielded overall 93.12% categorical agreement, 8.65% major errors, and 6.20% very major errors.
We implemented the classifier in Kleborate, and the Pathogenwatch web platform. Using this to assess the global distribution of ciprofloxacin resistance determinants in KpSC genomes available in Pathogenwatch (n=31,319, from 109 countries between years 2000-2023), we observed a significant positive association between national quinolone consumption rates and predicted ciprofloxacin resistance (R2=0.20, p=0.004).
All data, code, figures and tables are publicly available in this Github repository: https://github.com/klebgenomics/cipropaper
Full paper and details here.
Join us
Are you interested in joining the group and have matched Klebsiella pneumoniae species complex read/genome data with matched antibiotic susceptibility test data (minimum >100 genomes)?
As a member, you will be invited to be co-authors on these project outputs, provide feedback on interim results, edit manuscript drafts, test software updates, have the opportunity to propose analyses, and curate/review KpSC AMR rules as part of the ESGEM-AMR Klebsiella pneumoniae Working Group initiative.
Please contact Kara ([email protected]) for more details.
Membership
We already have over 100 members in our Genotype-Phenotype Group.
For more details about our members, please visit the AMRgenopheno Membership page.